
Ist Morbus Gaucher behandelbar?
Menschen mit lysosomalen Speicherkrankheiten sind nicht in der Lage, ausreichende Mengen bestimmter Enzyme für eine normale Körperfunktion herzustellen. Die Enzyme, die für den Abbau vieler verschiedener Stoffe erforderlich sind, sind bei den Betroffenen in zu geringer Menge oder überhaupt nicht vorhanden.
Das folgende Schaubild zeigt, was bei lysosomalen Speicherkrankheiten geschieht.
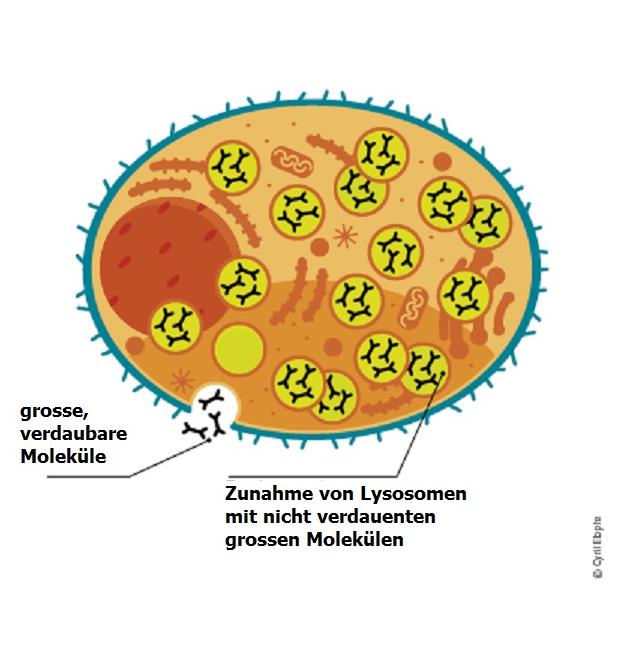
Es kommt zu einer Ansammlung von Stoffen, die die normale Zellfunktion stören. Dies kann zur Beeinträchtigung verschiedener Organe und zu lebensbedrohlichen Problemen führen.
Bei Morbus Gaucher ist der Körper nicht in der Lage, das Enzym Glukozerebrosidase herzustellen, das zur Ausscheidung bestimmter Abfallprodukte erforderlich ist. Forscher haben daher eine Behandlung namens Enzymersatztherapie (oder Enzymsubstitutionstherapie) entwickelt, bei der das fehlende Enzym im Körper ersetzt wird.
Enzymersatztherapie (EET)
Bei der EET oder auch ERT (engl. Enzyme replacement therapy) wird das fehlende lysosomale Enzym durch Infusionen eines Enzyms zugeführt, das der menschlichen Form sehr ähnlich ist.
Die Behandlung umfasst regelmässige Infusionen des fehlenden Enzyms alle zwei Wochen, um den Stoffwechsel zu normalisieren und die Ansammlung von Abfallstoffen zu verhindern. Die Infusionen werden allgemein gut vertragen und sind mit wenigen Nebenwirkungen verbunden.
Das folgende Diagramm erklärt die Wirkung der Enzymersatztherapie.
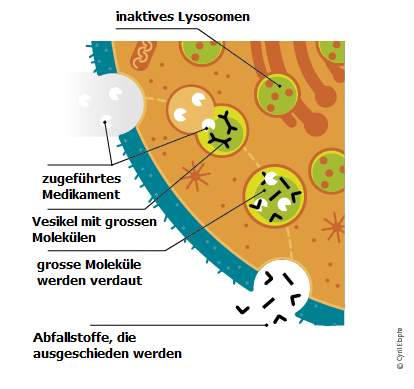
Es gibt zunehmend Hinweise darauf, dass die Therapie umso besser wirkt, je früher nach Einsetzen der ersten Symptome sie beginnt. Sie kann den Krankheitsverlauf verlangsamen und Spätfolgen vermeiden helfen; dadurch wird vielen Patienten ein normales Leben ermöglicht.
Bislang können die EET-Medikamente nicht die Blut-Hirn-Schranke passieren. Die EET hat daher wenig oder keinen Einfluss auf Symptome, die vom zentralen Nervensystem herrühren.
Die Enzymersatztherapie für Morbus Gaucher wurde seit ihrer Entwicklung weltweit bei mehr als 5.500 Patienten mit der Krankheit angewendet.
Auch für die Behandlung von der Fabry und Pompe Krankheit sowie der der Mukopolysaccharidose Typ 1 (MPS1 oder Hurler, Hurler-Scheie und Scheie Krankheit) steht eine Enzymersatztherapie zur Verfügung. Eine EET für zahlreiche andere lysosomale Speicherkrankheiten befindet sich derzeit noch in der Entwicklung.
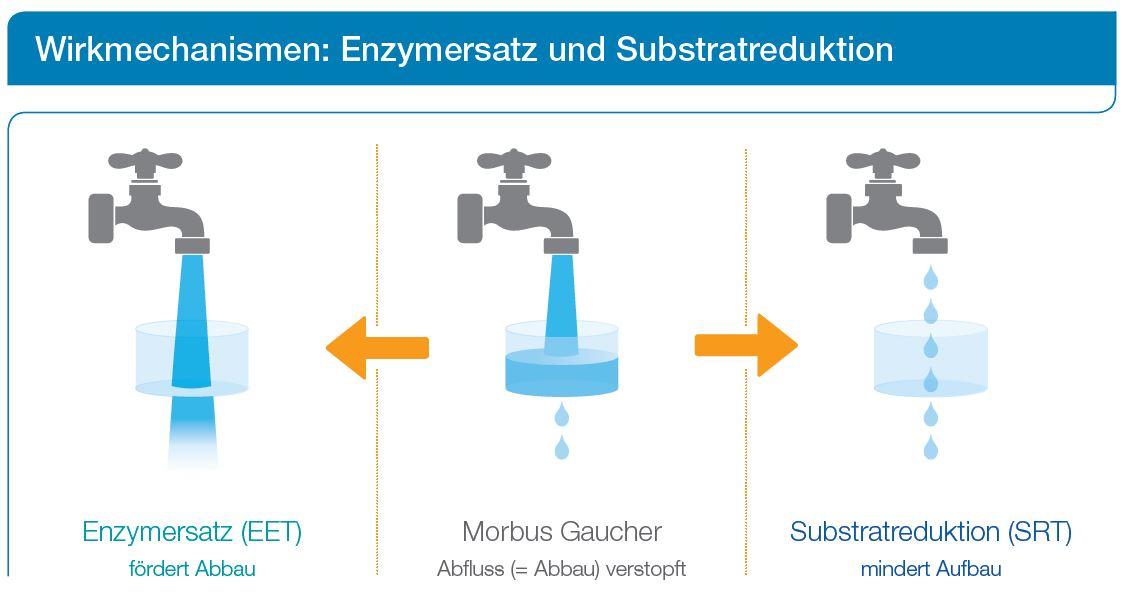
Substratreduktionstherapie (SRT)
Die Substratreduktionstherapie wird oral (d. h. über den Mund) verabreicht. Sie soll die Entstehung und Ansammlung von Abfallstoffen in den Zellen verringern.
Symptomlinderung
Es gibt eine Reihe von Therapien und Verfahren zur Linderung der Symptome von Morbus Gaucher. In der Vergangenheit wurde eine vergrößerte Milz oft operativ entfernt (Splenektomie). Knochenschmerzen können mithilfe von Schmerzmitteln behandelt werden, und bei defekter Hüfte ist ein Gelenkersatz möglich. All diese Behandlungsansätze zielen jedoch nicht spezifisch auf Morbus Gaucher ab. Sie bekämpfen nicht die zugrunde liegende Ursache der Krankheit und beheben nicht den Enzymmangel, der zur Ansammlung der Abfallstoffe führt.